singleRcapture: An R Package for Single-Source Capture-Recapture Models
Piotr Chlebicki, Maciej Beręsewicz
2025-02-13
Source:vignettes/singleRcapture.Rmd
singleRcapture.RmdIntroduction
Population size estimation is a methodological approach employed across multiple scientific disciplines, which serves as the basis for research, policy formulation, and decision-making processes (cf. Böhning, Bunge, and Heijden 2018). In the field of statistics, particularly official statistics, precise population estimates are essential in order to develop robust economic models, optimize resource allocation, and inform evidence-based policy (cf. Baffour, King, and Valente 2013). Social scientists utilize advanced population estimation techniques to investigate hard-to-reach populations, such as homeless individuals or illicit drug users in an effort to overcome the inherent limitations of conventional census methodologies. These techniques are crucial for obtaining accurate data on populations that are typically under-represented or difficult to access using traditional sampling methods (cf. Vincent and Thompson 2022). In ecology and epidemiology, researchers focus on estimating the size of individual species or disease-affected populations within defined geographical regions as part of conservation efforts, ecosystem management, and public health interventions.
Population size estimation can be approached using various methodologies, each with distinct advantages and limitations. Traditional approaches include full enumeration (e.g. censuses) and comprehensive sample surveys, which, while providing detailed data, are often resource-intensive and may result in delayed estimates, particularly for human populations. Alternative methods leverage existing data sources, such as administrative registers or carefully designed small-scale studies in wildlife research, or census coverage surveys (cf. Wolter 1986; Zhang 2019). Information from these sources is often extracted by applying statistical methods, known as capture-recapture or multiple system estimation, which rely on data from multiple enumerations of the same population (cf. Dunne and Zhang 2024). This approach can be implemented using either a single source with repeated observations, two sources, or multiple sources.
In this paper we focus on methods that involve a single data source with multiple enumerations of the same units (cf. Heijden et al. 2003). In human population studies, such data can be derived from police records, health system databases, or border control logs; in the case of non-human populations, data of this kind can come from veterinary records or specialized field data. These methods are often applied to estimate hard-to-reach or hidden populations, where standard sampling methods may be inappropriate because of prohibitive costs or problems with identifying population members.
While methods for two or more sources are implemented in various
open-source software packages, for instance CARE-4
(Yang and Chao
2006) (in GAUSS), Rcapture
(Baillargeon and Rivest
2007), marked (Laake et al. 2013) or
VGAM (Thomas W. Yee, Stoklosa, and Huggins
2015) (all in R), single-source
capture-recapture (SSCR) methods are either not available at all or are
only partially implemented in existing R packages or
other software. Therefore, the paper attempts to bridge this gap by
introducing the singleRcapture package, which implement
state-of-the-art SSCR methods and offer a user friendly API
resembling existing R functions (e.g.,
glm). In the next subsection we describe existing
R packages and other software that could be used for
estimating population size using SSCR methods.
Software for capture-recapture with a single source
The majority of SSCR methods assume zero-truncated distributions or
their extensions (e.g., inclusion of one-inflation). The
Distributions.jl (Besançon et al. 2021) (in
Julia), StatsModels (Seabold and
Perktold 2010) (in Python),
countreg (Zeileis, Kleiber, and Jackman 2008),
VGAM (T. Yee 2015) or
distributions3 (Hayes et al. 2024) (in
R) implement some of those truncated distributions
(e.g. distributions3::ZTPoisson or
countreg::zerotrunc) and the most general distributions,
such as Generally Altered, Inflated, Truncated and Deflated, can be
found in the VGAM package (e.g.
VGAM::gaitdpoisson for the Poisson distribution, see Thomas W. Yee and Ma (2024) for a detailed description).
However, the estimation of parameters of a given truncated (and possibly
inflated) distribution is just the first step (as in the case of
log-linear models in capture-recapture with two sources) and, to the
best of our knowledge, there is no open-source software that can be used
to estimate population size using on SSCR methods and includes variance
estimators or diagnostics.
Therefore, the purpose of the singleRcapture, an R package, is to bridge this gap by providing scientists and other practitioners with a tool for estimating population size with SSCR methods. We have implemented state-of-the-art methods, as recently described by Böhning, Bunge, and Heijden (2018) or Böhning and Friedl (2024) and provided their extensions (e.g., inclusion of covariates, different treatment of one-inflation), which will be covered in detail in Section 2. The package implements variance estimation based on various methods, can be used to create custom models and diagnostic plots (e.g. rootograms) with parameters estimated using a modified iteratively reweighted least squares (IRLS) algorithm we have implemented for estimation stability. To the best of our knowledge, no existing open-source package allows the estimation of population size by selecting an appropriate SSCR model, conducting the estimation, and providing informative diagnostics of the results.
The remaining part of the paper is structured as follows. Section 2
contains a brief description of the theoretical background and
information about fitting methods and available methods of variance
estimation. Section 3 introduces the main functions of the package.
Section 4 presents a case study and contains an assessment of its
results, diagnostics and estimates of specific sub-populations. Section
5 describes classes and S3methods implemented in the
package. The paper ends with conclusions and an appendix showing how to
use the estimatePopsizeFit function which is aimed to mimic
the glm.fit or similar functions. In replication materials
we show how to implement a custom model as this option could be of
interest to users wishing to apply any new bootstrap methods not
implemented in the package.
Theoretical background
How to estimate population size with a single source?
Let represent the number of times the -th unit was observed in a single source (e.g. register). Clearly, we only observe and do not know how many units have been missed (i.e. ), so the population size, denoted by , needs to be estimated. In general, we assume that the conditional distribution of given a vector of covariates follows a version of the zero-truncated count data distribution (and its extensions). When we know the parameters of the distribution we can estimate the population size using a Horvitz-Thompson type estimator given by:
where , is the number of observed units and is the indicator function, while the maximum likelihood estimate of is obtained after substituting regression parameters for in the above equation.
The basic SSCR assumes independence between counts, which is a rather naive assumption, since the first capture may significantly influence the behavior of a given unit or limit the possibility of subsequent captures (e.g. due to incarceration).
To solve these issues, Ryan T. Godwin and Böhning (2017b) and Ryan T. Godwin and Böhning (2017a) introduced one-inflated distributions, which explicitly model the probability of singletons by giving additional mass to singletons denoted as (cf. Böhning and Friedl 2024). In other words they considered a new random varialbe that corresponds to the data collection process which exhibits one inflation:
Analytic variance estimation is then performed by computing two parts of the decomposition according to the law of total variance given by:
where the first part can be estimated using the multivariate method given by:
while the second part of the decomposition above, assuming independence of ’s and after some omitted simplifications, is optimally estimated by:
which serves as the basis for interval estimation. Confidence intervals are usually constructed under the assumption of (asymptotic) normality of or asymptotic normality of (or log normality of ). The latter is an attempt to address a common criticism of student type confidence intervals in SSCR, namely a possibly skewed distribution of , and results in the confidence interval given by:
where:
and where is the quantile function of the standard normal distribution. The estimator is best interpreted as being an estimator of the total number of units in the population, since we have no means of estimating the number of units in the population for which the probability of being included in the data is (Heijden et al. 2003).
Available models
The full list of models implemented in singleRcapture along with corresponding expressions for probability density functions and point estimates can be found in the collective help file for all family functions:
?ztpoissonFor the sake of simplicity, we only list the family functions together with brief descriptions. For more detailed information, please consult the relevant literature.
The current list of these family functions includes:
- Generalized Chao’s (Chao 1987) and Zelterman’s (Zelterman 1988) estimators via logistic regression on variable defined as if and if with where is the Bernoulli distribution and can be modeled for each unit by with Poisson parameter (for a covariate extension see Böhning et al. (2013) and Böhning and Heijden (2009)):
where and denotes number of units observed once and twice.
- Zero-truncated
(
zt) and zero-one-truncated (zot) Poisson (Böhning and Heijden 2019), geometric, NB type II (NB2) regression, where the non-truncated distribution is parameterized as:
- Zero-truncated one-inflated
(
ztoi) modifications, where the count data variable is defined such that its distribution statisfies:
- One-inflated zero-truncated
(
oizt) modifications, where the count data variable is defined as:
Note that
ztoi
and
oizt
distributions are equivalent, in the sense that the maximum value of the
likelihood function is equal for both of those distributions given any
data, as shown by (Böhning 2023) but
population size estimators are different.
In addition, we propose two new approaches to model singletons in a similar way as in hurdle models. In particular, we have proposed the following:
- The zero-truncated hurdle model (
ztHurdle*) for Poisson, geometric and NB2 is defined as:
where denotes the conditional probability of observing singletons.
- The hurdle zero-truncated model (
Hurdlezt*) for Poisson, geometric and NB2 is defined as:
where denotes the unconditional probability of observing singletons.
The approaches presented above differ in their assumptions,
computational complexity, or in the way they treat heterogeneity of
captures and singletons. For instance, the dispersion parameter
in the NB2 type models is often interpreted as measuring the
severity of unobserved heterogeneity in the underlying
Poisson process (Cruyff
and Heijden 2008). When using any truncated NB model, the
hope is that given the class of models considered, the consistency is
not lost despite the lack of information.
While not discussed in the literature, the interpretation of
heterogeneous
across the population (specified in controlModel) would be
that the unobserved heterogeneity affects the accuracy of the prediction
for the dependent variable
more severely than others. The geometric model (NB with
)
is singled out in the package and often considered in the literature
because of inherent computational issues with NB models, which are
exacerbated by the fact that data used for SSCR are usually of rather
low quality. Data sparsity is a particularly common problem in SSCR and
a big challenge for all numerical methods for fitting the
(zero-truncated) NB model.
The extra mass
in one-inflated models is an important extension to the researcher’s
toolbox for SSCR models, since the inflation at
is likely to occur in many types of applications. For example, when
estimating the number of active people who committed criminal acts in a
given time period, the fact of being captured for the first time
following an arrest is associated with the risk of no longer being able
to be captured a second time. One constraint present in modelling via
inflated models is that attempts to include both the possibility of one
inflation and one deflation lead to both numerical and inferential
problems since the parameter space (of
or
)
is then given by
for the probability mass function
.
The boundary of this set is then a
or
dimentional
manifold, transforming this parameter space into
would require using link functions that depend on more than
one parameter.
Hurdle models represent another approach to modelling one-inflation. They can also model deflation as well as inflation and deflation simultaneously, so they are more flexible and, in the case of hurdle zero-truncated models, appear to be more numerically stable.
Although the question of how to interpret regression parameters tends
to be somewhat overlooked in SSCR studies, we should point out that the
interpretation of the
inflation parameter (in
ztoi
or
oizt)
is more convenient than the interpretation of the
probability parameter (in hurdle models). Additionally, the
interpretation of the
parameter in (one) inflated models conforms to the following intuition:
given that unit
comes from the non-inflated part of the population, it follows a Poisson
distribution (respectively geometric or negative binomial) with the
parameter (or
);
no such interpretation exists for hurdle models. Interestingly,
estimates from hurdle zero-truncated and one-inflated zero-truncated
models tend to be quite close to one another, although more rigorous
studies are required to confirm this observation.
Fitting method
As previously noted, the singleRcapture package can
be used to model the (linear) dependence of all parameters on
covariates. A modified IRLS algorithm is employed for this purpose as
presented in Algorithm 1; full details are available in T. Yee (2015). In
order to apply the algorithm, a modified model matrix
is created when the estimatePopsize function is called. In
the context of the models implemented in
singleRcapture, this matrix can be written as:
where each corresponds to a model matrix associated with a user specified formula.
Algorithm 1: The modified IRLS algorithm used in the singleRcapture package
Initialize with
iter← 1, ←start, ← I, ←-
Store values from the previous step:
← , ← , ← (the last assignment is omitted during the first iteration), and assign values in the current iteration:←
←
←
where denotes offset.
Assign the current coefficient value:
←If try selecting the smallest value such that for ← the inequality holds if this is successful ← , else stop the algorithm.
If convergence is achieved or
iteris higher thanmaxiter, stop the algorithm, elseiter← 1 +iterand return to step 2.
In the case of multi-parameter families, we get a matrix of linear predictors instead of a vector, with the number of columns matching the number of parameters in the distribution. “Weights” (matrix ) are then modified to be information matrices , where is the log-likelihood function and is the -th row of , while in the typical IRLS they are scalars , which is often just .
Bootstrap variance estimators
We have implemented three types of bootstrap algorithms: parametric (adapted from theory in Zwane and Van der Heijden (2003), Norris and Pollock (1996) for multiple source setting with covariates), semi-parametric (see e.g. Böhning and Friedl (2021)) and nonparametric. The nonparametric version is the usual bootstrap algorithm; which will typically underestimate the variance of . In this section, the focus is on the first two approaches.
The idea of semi-parametric bootstrap is to modify the usual bootstrap to include the additional uncertainty resulting from the fact that the sample size is a random variable. This type of bootstrap is performed in steps listed in Algorithm 2.
Algorithm 2: Semi-parametric bootstrap
- Draw a sample of size , where
- Draw units from the data uniformly without replacement
- Obtain a new population size estimate using bootstrap data
- Repeat 1-3 steps times
In other words, we first draw a sample size and then a sample conditional on the sample size. Note that when using the semi-parametric bootstrap one implicitly assumes that the population size estimate is accurate. The last implemented bootstrap type is the parametric algorithm, which first draws a finite population of size from the superpopulation model and then samples from this population according to the selected model, as described in Algorithm 3.
Algorithm 3: Parametric bootstrap
- Draw the number of covariates equal to proportional to the estimated contribution with replacement
- Using the fitted model and regression coefficients draw for each covariate the value from the corresponding probability measure on
- Truncate units with the drawn value equal to
- Obtain a population size estimate based on the truncated data
- Repeat 1-4 steps times
Note that in order for this type of algorithm to result in consistent standard error estimates, it is imperative that the estimated model for the entire superpopulation probability space is consistent, which may be much less realistic than in the case of the semi-parametric bootstrap. The parametric bootstrap algorithm is the default option in singleRcapture.
The main function
The estimatePopsize function
The singleRcapture package is built around the
estimatePopsize function. The main design objective was to
make using estimatePopsize as similar as possible to the
standard stats::glm function or packages for fitting
zero-truncated regression models, such as countreg
(e.g. countreg::zerotrunc function). The
estimatePopsize function is used to first fit an
appropriate (vector) generalized linear model and to estimate the
population size along with its variance. It is assumed that the response
vector (i.e. the dependent variable) corresponds to the number of times
a given unit was observed in the source. The most important arguments
are given in Table below; the obligatory ones are
formula, data, model.
| Argument | Description |
|---|---|
formula |
The main formula (i.e., for the Poisson parameter); |
data |
A data.frame (or data.frame coercible)
object; |
model |
Either a function, a string, or a family class object specifying
which model should be used; possible values are listed in the
documentation. The supplied argument should have the form
model = "ztpoisson", model = ztpoisson, or if
a link function should be specified, then
model = ztpoisson(lambdaLink = "log") can be used; |
method |
A numerical method used to fit regression IRLS or
optim; |
popVar |
A method for estimating variance of and creating confidence intervals (either bootstrap, analytic, or skipping the estimation entirely); |
controlMethod, controlModel, or
controlPopVar
|
Control parameters for numerical fitting, specifying additional formulas (inflation, dispersion) and population size estimation, respectively; |
offset |
A matrix of offset values with the number of columns matching the number of distribution parameters, providing offset values to each of the linear predictors; |
... |
Additional optional arguments passed to other methods, e.g.,
estimatePopsizeFit; |
An important step in using estimatePopsize is specifying
the model parameter, which indicates the type of model that
will be used for estimating the unobserved part of the
population. For instance, to fit Chao’s or Zelterman’s model one should
select chao or zelterman and, assuming that
one-inflation is present, one can select one of the zero-truncated
one-inflated
(ztoi)
or one-inflated zero-truncated
(oizt)
models, such as oiztpoisson for Poisson or
ztoinegbin for NB2.
If it is assumed that heterogeneity is observed for NB2 models, one
can specify the formula in the controlModel argument with
the controlModel function and the alphaFormula
argument. This enables the user to provide a formula for the dispersion
parameter in the NB2 models. If heterogeneity is assumed for
ztoi
or
oizt,
one can specify the omegaFormula argument, which
corresponds to the
parameter in these models. Finally, if covariates are assumed to be
available for the hurdle models
(ztHurdle
or
Hurdlezt),
then piFormula can be specified, as it provides a formula
for the probability parameter in these models.
Controlling variance estimation with controlPopVar
The estimatePopsize function makes it possible to
specify the variance estimation method via popVar
(e.g. analytic or variance bootstrap) and control the estimation process
by specifying controlPopVar. In the control function
controlPopVar the user can specify the
bootType argument, which has three possible values:
"parametric", "semiparametric" and
"nonparametric". Additional arguments accepted by the
contorlPopVar function, which are relevant to bootstrap,
include:
-
alpha,B– the significance level and the number of bootstrap samples to be performed, respectively, with and being the default options. -
cores– the number of process cores to be used in bootstrap (1 by default); parallel computing is enabled by doParallel (Microsoft and Weston 2022a), foreach (Microsoft and Weston 2022b) and parallel packages (R Core Team 2023). -
keepbootStat– a logical value indicating whether to keep a vector of statistics produced by the bootstrap. -
traceBootstrapSize,bootstrapVisualTrace– logical values indicating whether sample and population size should be tracked (FALSEby default); these work only whencores= 1. -
fittingMethod,bootstrapFitcontrol– the fitting method (by default the same as the one used in the original call) and control parameters (controlMethod) for model fitting in the bootstrap.
In addition, the user can specify the type of confidence interval by
means of confType and the type of covariance matrix by
using covType for the analytical variance estimator
(observed or the Fisher information matrix).
In the next sections we present a case study involving the use of a
simple zero-truncated Poisson regression and a more advanced model:
one-inflated zero-truncated geometric regression with the
cloglog link function. First, we present the example
dataset, then we describe how to estimate the population size and assess
the quality and diagnostics measures. Finally, we show how to estimate
the population size in user-specified sub-populations.
Data analysis example
The package can be installed in the standard manner using:
install.packages("singleRcapture")Then, we need to load the package using the following code:
Dataset
We use a dataset from Heijden et al. (2003), which contains information about
immigrants in four Dutch cities (Amsterdam, Rotterdam, The Hague and
Utrecht), who were staying in the country without a legal permit in 1995
and appeared in police records for that year. This dataset is included
in the package called netherlandsimmigrant:
## capture gender age reason nation
## 1 1 male <40yrs Other reason North Africa
## 2 1 male <40yrs Other reason North Africa
## 3 1 male <40yrs Other reason North Africa
## 4 1 male <40yrs Other reason Asia
## 5 1 male <40yrs Other reason Asia
## 6 2 male <40yrs Other reason North AfricaThe number of times each individual appeared in the records is
included in the capture variable. The available covariates
include gender, age, reason, nation; the last two represent
the reason for being captured and the region of the world a given person
comes from:
summary(netherlandsimmigrant)## capture gender age reason
## Min. :1.000 female: 398 <40yrs:1769 Illegal stay: 259
## 1st Qu.:1.000 male :1482 >40yrs: 111 Other reason:1621
## Median :1.000
## Mean :1.162
## 3rd Qu.:1.000
## Max. :6.000
## nation
## American and Australia: 173
## Asia : 284
## North Africa :1023
## Rest of Africa : 243
## Surinam : 64
## Turkey : 93One notable characteristic of this dataset is that it contains a disproportionately large number of individuals who were observed only once (i.e. 1645).
table(netherlandsimmigrant$capture)##
## 1 2 3 4 5 6
## 1645 183 37 13 1 1The basic syntax of estimatePopsize is very similar to
that of glm, the same can be said about the output of the
summary method except for additional results of population size
estimates (denoted as
Population size estimation results).
basicModel <- estimatePopsize(
formula = capture ~ gender + age + nation,
model = ztpoisson(),
data = netherlandsimmigrant,
controlMethod = controlMethod(silent = TRUE)
)
summary(basicModel)##
## Call:
## estimatePopsize.default(formula = capture ~ gender + age + nation,
## data = netherlandsimmigrant, model = ztpoisson(), controlMethod = controlMethod(silent = TRUE))
##
## Pearson Residuals:
## Min. 1st Qu. Median Mean 3rd Qu. Max.
## -0.486442 -0.486442 -0.298080 0.002093 -0.209444 13.910844
##
## Coefficients:
## -----------------------
## For linear predictors associated with: lambda
## Estimate Std. Error z value P(>|z|)
## (Intercept) -1.3411 0.2149 -6.241 4.35e-10 ***
## gendermale 0.3972 0.1630 2.436 0.014832 *
## age>40yrs -0.9746 0.4082 -2.387 0.016972 *
## nationAsia -1.0926 0.3016 -3.622 0.000292 ***
## nationNorth Africa 0.1900 0.1940 0.979 0.327398
## nationRest of Africa -0.9106 0.3008 -3.027 0.002468 **
## nationSurinam -2.3364 1.0136 -2.305 0.021159 *
## nationTurkey -1.6754 0.6028 -2.779 0.005445 **
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
##
## AIC: 1712.901
## BIC: 1757.213
## Residual deviance: 1128.553
##
## Log-likelihood: -848.4504 on 1872 Degrees of freedom
## Number of iterations: 8
## -----------------------
## Population size estimation results:
## Point estimate 12690.35
## Observed proportion: 14.8% (N obs = 1880)
## Std. Error 2808.169
## 95% CI for the population size:
## lowerBound upperBound
## normal 7186.444 18194.26
## logNormal 8431.275 19718.32
## 95% CI for the share of observed population:
## lowerBound upperBound
## normal 10.332927 26.16037
## logNormal 9.534281 22.29793The output regarding the population size contains the point estimate, the observed proportion (based on the input dataset), the standard error and two confidence intervals: one relating to the point estimate, the second – to the observed proportion.
According to this simple model, the population size is about 12,500, with about 15% of units observed in the register. The 95% CI under normality indicates that the true population size is likely to be between 7,000-18,000, with about 10-26% of the target population observed in the register.
Since there is a reasonable suspicion that the act of observing a
unit in the dataset may lead to undesirable consequences for the person
concerned (in this case, a possible deportation, detention or something
similar). For these reasons, the user may consider one-inflated models,
such as one-inflated zero-truncated geometric model (specified by
oiztgeom family) and those presented below.
set.seed(123456)
modelInflated <- estimatePopsize(
formula = capture ~ nation,
model = oiztgeom(omegaLink = "cloglog"),
data = netherlandsimmigrant,
controlModel = controlModel(
omegaFormula = ~ gender + age
),
popVar = "bootstrap",
controlPopVar = controlPopVar(bootType = "semiparametric",
B = 50)
)
summary(modelInflated)##
## Call:
## estimatePopsize.default(formula = capture ~ nation, data = netherlandsimmigrant,
## model = oiztgeom(omegaLink = "cloglog"), popVar = "bootstrap",
## controlModel = controlModel(omegaFormula = ~gender + age),
## controlPopVar = controlPopVar(bootType = "semiparametric",
## B = 50))
##
## Pearson Residuals:
## Min. 1st Qu. Median Mean 3rd Qu. Max.
## -0.41643 -0.41643 -0.30127 0.00314 -0.18323 13.88376
##
## Coefficients:
## -----------------------
## For linear predictors associated with: lambda
## Estimate Std. Error z value P(>|z|)
## (Intercept) -1.2552 0.2149 -5.840 5.22e-09 ***
## nationAsia -0.8193 0.2544 -3.220 0.00128 **
## nationNorth Africa 0.2057 0.1838 1.119 0.26309
## nationRest of Africa -0.6692 0.2548 -2.627 0.00862 **
## nationSurinam -1.5205 0.6271 -2.425 0.01532 *
## nationTurkey -1.1888 0.4343 -2.737 0.00619 **
## -----------------------
## For linear predictors associated with: omega
## Estimate Std. Error z value P(>|z|)
## (Intercept) -1.4577 0.3884 -3.753 0.000175 ***
## gendermale -0.8738 0.3602 -2.426 0.015267 *
## age>40yrs 1.1745 0.5423 2.166 0.030326 *
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
##
## AIC: 1677.125
## BIC: 1726.976
## Residual deviance: 941.5416
##
## Log-likelihood: -829.5625 on 3751 Degrees of freedom
## Number of iterations: 10
## -----------------------
## Population size estimation results:
## Point estimate 6699.953
## Observed proportion: 28.1% (N obs = 1880)
## Boostrap sample skewness: 0.5209689
## 0 skewness is expected for normally distributed variable
## ---
## Bootstrap Std. Error 1579.235
## 95% CI for the population size:
## lowerBound upperBound
## 5107.533 10738.242
## 95% CI for the share of observed population:
## lowerBound upperBound
## 17.50752 36.80838According to this approach, the population size is about 7,000, which
is about 5,000 less than in the case of the naive Poisson approach. A
comparison of AIC and BIC suggests that the one-inflation model fits the
data better with BIC for oiztgeom 1727 and 1757 for
ztpoisson.
We can access population size estimates using the following code, which returns a list with numerical results.
popSizeEst(basicModel) # alternative: basicModel$populationSize## Point estimate: 12690.35
## Variance: 7885812
## 95% confidence intervals:
## lowerBound upperBound
## normal 7186.444 18194.26
## logNormal 8431.275 19718.32
popSizeEst(modelInflated) # alternative: modelInflated$populationSize## Point estimate: 6699.953
## Variance: 2493985
## 95% confidence intervals:
## lowerBound upperBound
## 5107.533 10738.242The decision whether to use a zero-truncated Poisson or one-inflated zero-truncated geometric model should be based on the assessment of the model and the assumptions regarding the data generation process. One possible method of selection is based on the likelihood ratio test, which can be computed quickly and conveniently with the lmtest (Zeileis and Hothorn (2002)) interface:
library(lmtest)
lrtest(basicModel, modelInflated,
name = function(x) {
if (family(x)$family == "ztpoisson")
"Basic model"
else "Inflated model"
})## Likelihood ratio test
##
## Model 1: Basic model
## Model 2: Inflated model
## #Df LogLik Df Chisq Pr(>Chisq)
## 1 8 -848.45
## 2 9 -829.56 1 37.776 7.936e-10 ***
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1However, the above is not a standard method of model selection in SSCR. The next sections are dedicated to a detailed description of how to assess the results using standard statistical tests and diagnostics.
Testing marginal frequencies
A popular method of testing the model fit in single source capture-recapture studies consists in comparing the fitted marginal frequencies with the observed marginal frequencies for . If the fitted model bears sufficient resemblance to the real data collection process, these quantities should be quite close and both and tests can be used to test the statistical significance of the discrepancy with the following singleRcapture syntax for the Poisson model (rather poor fit):
margFreq <- marginalFreq(basicModel)
summary(margFreq, df = 1, dropl5 = "group")## Test for Goodness of fit of a regression model:
##
## Test statistics df P(>X^2)
## Chi-squared test 50.06 1 1.5e-12
## G-test 34.31 1 4.7e-09
##
## --------------------------------------------------------------
## Cells with fitted frequencies of < 5 have been grouped
## Names of cells used in calculating test(s) statistic: 1 2 3and for the one-inflated model (better fit):
margFreq_inf <- marginalFreq(modelInflated)
summary(margFreq_inf, df = 1, dropl5 = "group")## Test for Goodness of fit of a regression model:
##
## Test statistics df P(>X^2)
## Chi-squared test 1.88 1 0.17
## G-test 2.32 1 0.13
##
## --------------------------------------------------------------
## Cells with fitted frequencies of < 5 have been grouped
## Names of cells used in calculating test(s) statistic: 1 2 3 4where the dropl5 argument is used to indicate how to
handle cells with less than
fitted observations. Note, however, that currently there is no
continuity correction.
Diagnostics
The singleRStaticCountData class has a plot
method implementing several types of quick demonstrative plots, such as
the rootogram (Kleiber
and Zeileis 2016), for comparing fitted and marginal
frequencies, which can be generated with the following syntax:
plot( basicModel, plotType = "rootogram", main = "ZT Poisson model")
plot(modelInflated, plotType = "rootogram", main = "OI ZT Geometric model")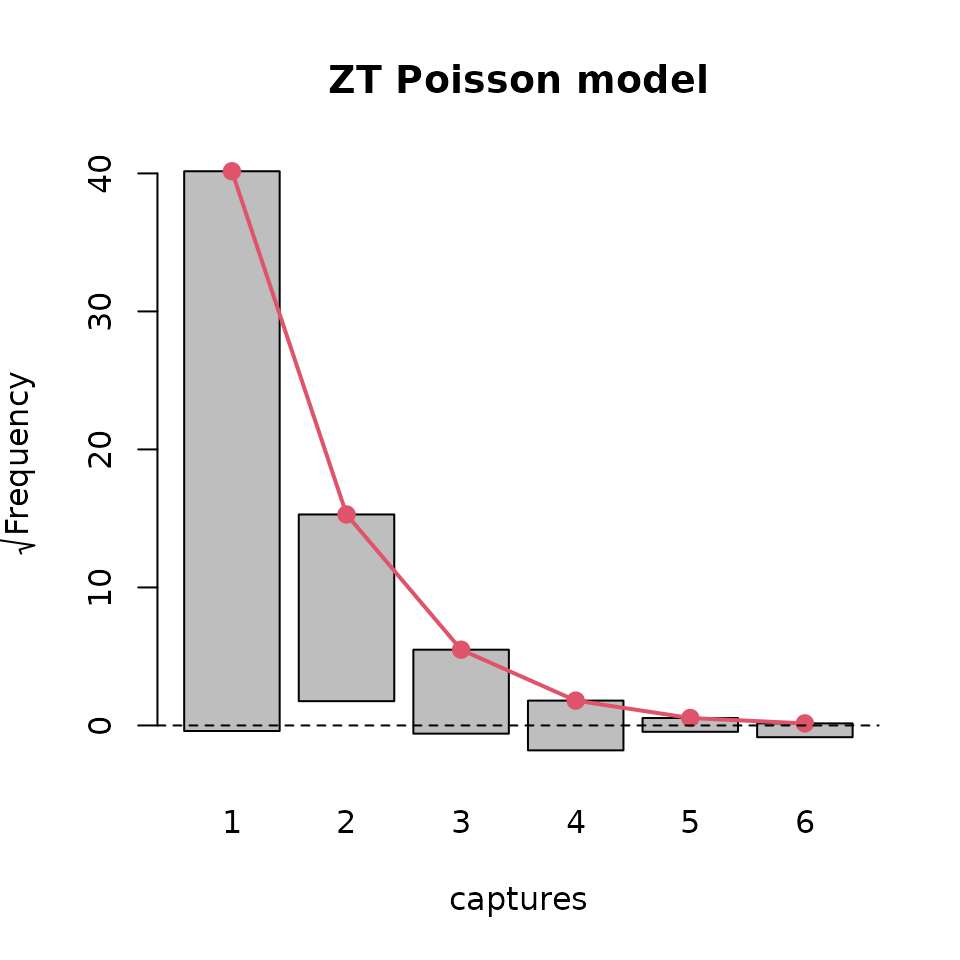
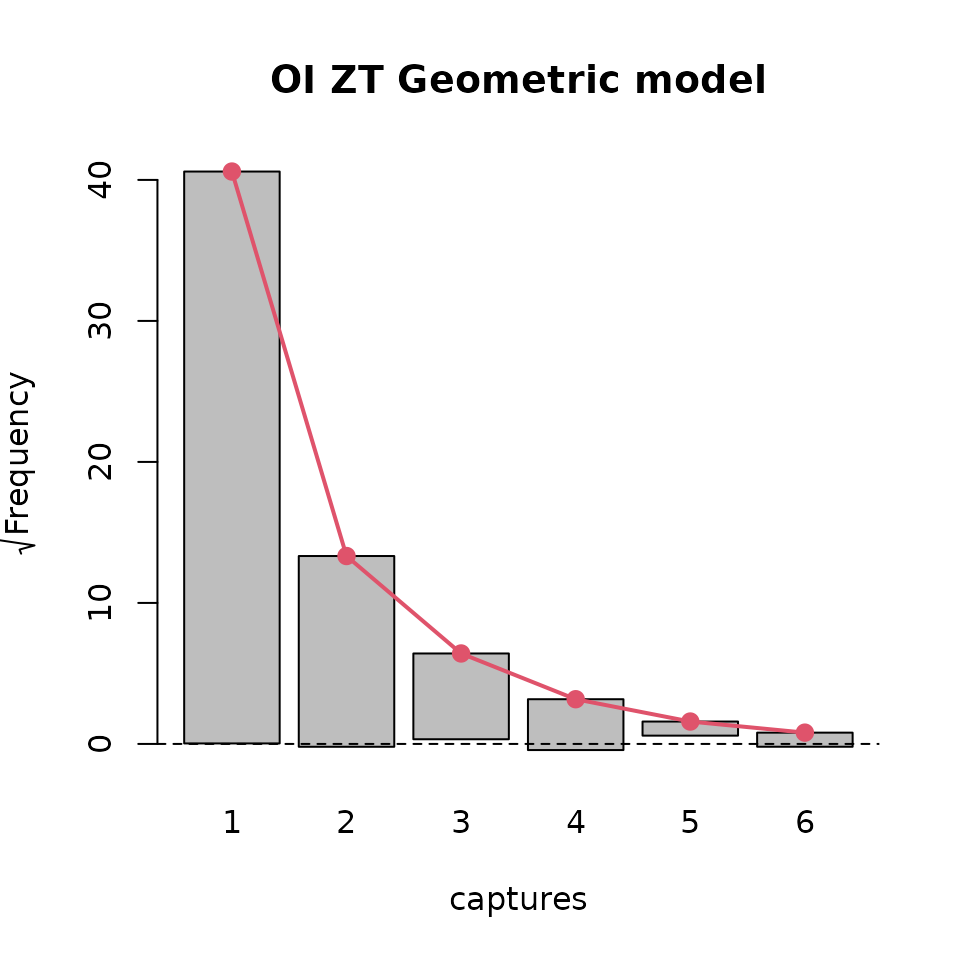
Rootograms for ztpoisson (left) and oiztgeom (right) models
The above plots suggest that the oiztgeom model fits the
data better. Another important issue in population size estimation is to
conduct model diagnostics in order to verify whether influential
observations are present in the data. For this purpose the leave-one-out
(LOO) diagnostic implemented in the dfbeta from the
stats package has been adapted as shown below
(multiplied by a factor of a hundred for better readability):
## 0% 25% 50% 75% 100%
## (Intercept) -0.9909 -0.1533 0.0191 0.0521 8.6619
## gendermale -9.0535 -0.0777 -0.0283 0.1017 2.2135
## age>40yrs -2.0010 0.0179 0.0379 0.0691 16.0061
## nationAsia -9.5559 -0.0529 0.0066 0.0120 17.9914
## nationNorth Africa -9.6605 -0.0842 -0.0177 0.0087 3.1260
## nationRest of Africa -9.4497 -0.0244 0.0030 0.0083 10.9787
## nationSurinam -9.3140 -0.0066 0.0020 0.0035 99.3383
## nationTurkey -9.6198 -0.0220 0.0079 0.0143 32.0980## 0% 25% 50% 75% 100%
## (Intercept) -1.4640 0.0050 0.0184 0.0557 9.0600
## nationAsia -6.6331 -0.0346 0.0157 0.0347 12.2406
## nationNorth Africa -7.2770 -0.0768 -0.0170 0.0085 1.9415
## nationRest of Africa -6.6568 -0.0230 0.0081 0.0262 7.1710
## nationSurinam -6.2308 -0.0124 0.0162 0.0421 62.2045
## nationTurkey -6.4795 -0.0273 0.0204 0.0462 21.1338
## (Intercept):omega -6.8668 -0.0193 0.0476 0.0476 9.3389
## gendermale:omega -2.2733 -0.2227 0.1313 0.2482 11.1234
## age>40yrs:omega -30.2130 -0.2247 -0.1312 -0.0663 2.0393The result of the dfbeta can be further used in the
dfpopsize function, which can be used to quantify LOO on
the population size. Note the warning when the bootstap variance
estimation is applied.
dfb_pop <- dfpopsize(basicModel, dfbeta = dfb)
dfi_pop <- dfpopsize(modelInflated, dfbeta = dfi)
summary(dfb_pop)## Min. 1st Qu. Median Mean 3rd Qu. Max.
## -4236.412 2.664 2.664 5.448 17.284 117.448
summary(dfi_pop)## Min. 1st Qu. Median Mean 3rd Qu. Max.
## -456.6443 -3.1121 -0.7243 3.4333 5.1535 103.5949Figure 2 shows a comparison of the effect of deleting an observation on the population size estimate and inverse probability weights, which refer to the contribution of a given observation to the population size estimate:
plot(basicModel, plotType = "dfpopContr",
dfpop = dfb_pop, xlim = c(-4500, 150))
plot(modelInflated, plotType = "dfpopContr",
dfpop = dfi_pop, xlim = c(-4500, 150))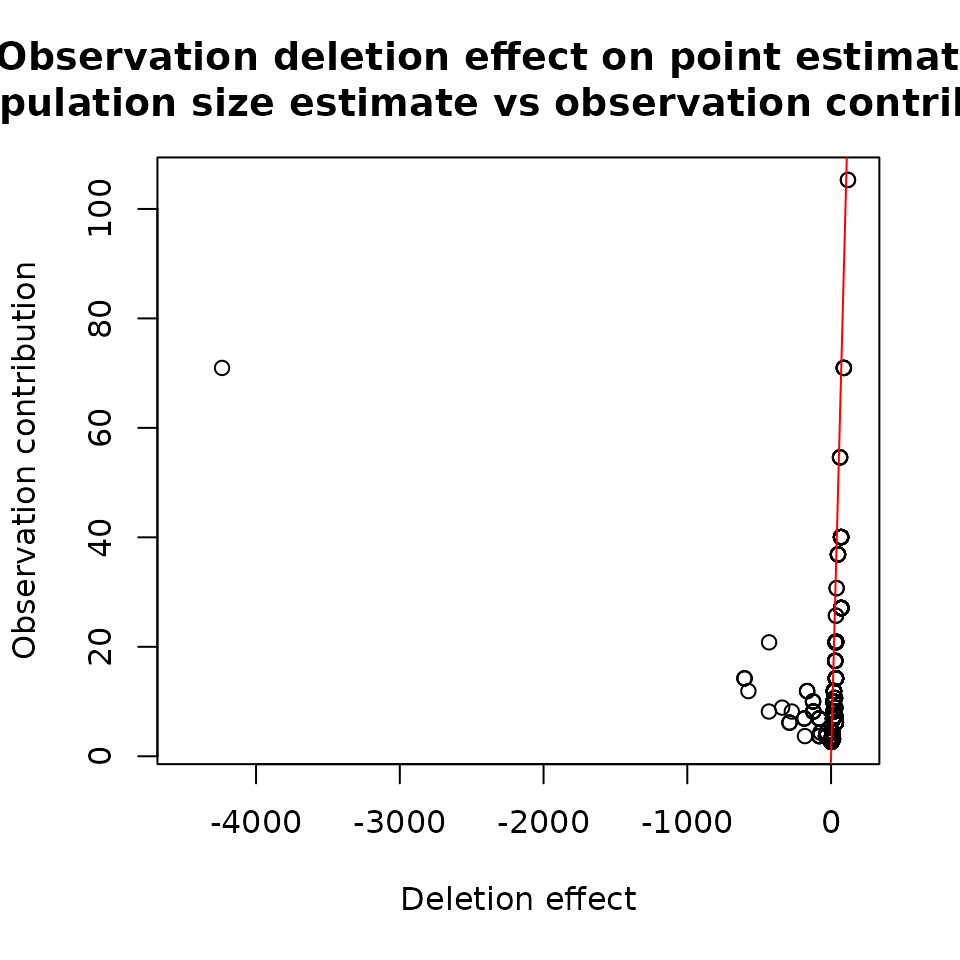
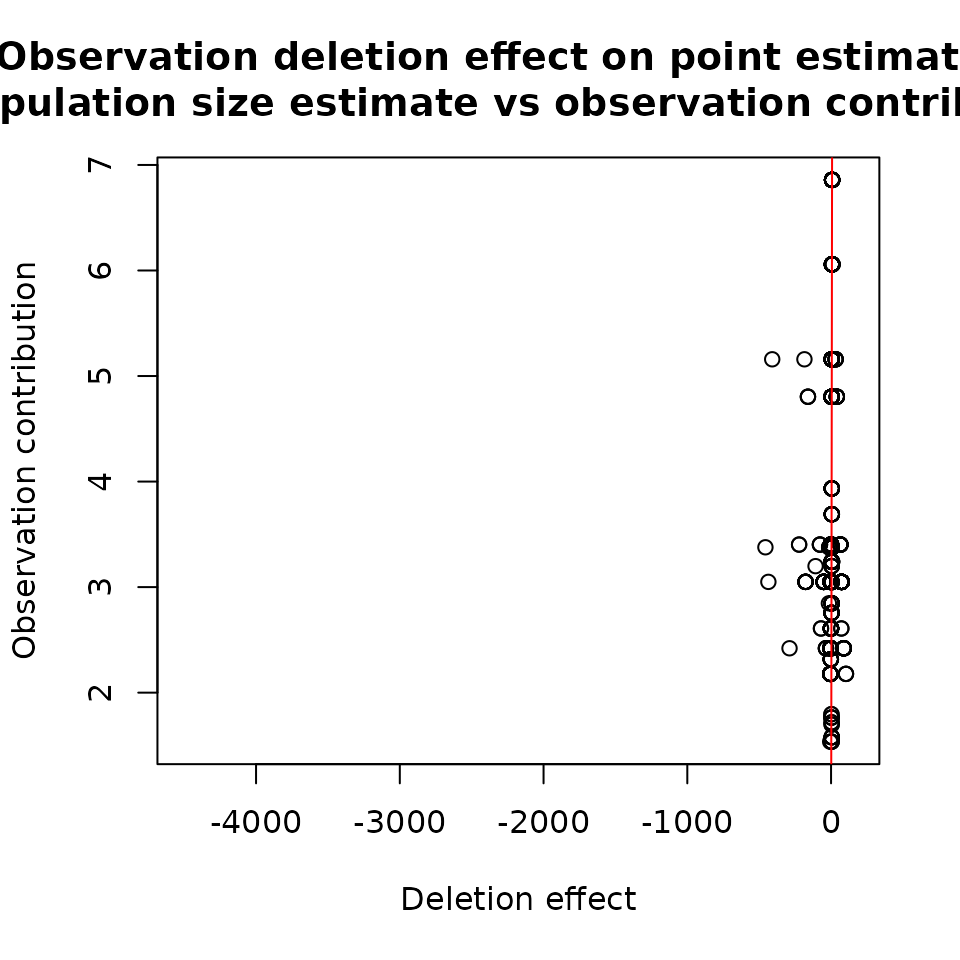
Results for ztpoisson (left) and oiztgeom (right) model
These plots show how the population size changes if a given
observation is removed. For instance, if we remove observation 542, then
the population size will increase by about 4236 for the
ztpoisson model. In the case of oiztgeom, the
largest change is equal to 457 for observation 900.
The full list of plot types along with the list of optional arguments
that can be passed from the call to the plot method down to
base R and graphics functions can be
found in the help file of the plot method.
?plot.singleRStaticCountDataThe stratifyPopsize function
Researchers may be interested not only in the total population size
but also in the size of specific sub-populations (e.g. males, females,
particular age groups). For this reason we have created the
stratifyPopsize function, which estimates the size by
strata defined by the coefficients in the model (the default option).
The following output presents results based on the
ztpoisson and oiztgeom models.
popSizestrata <- stratifyPopsize(basicModel)
cols <- c("name", "Observed", "Estimated", "logNormalLowerBound",
"logNormalUpperBound")
popSizestrata_report <- popSizestrata[, cols]
cols_custom <- c("Name", "Obs", "Estimated", "LowerBound", "UpperBound")
names(popSizestrata_report) <- cols_custom
popSizestrata_report## Name Obs Estimated LowerBound UpperBound
## 1 gender==female 398 3811.0924 2189.0439 6902.140
## 2 gender==male 1482 8879.2613 6090.7752 13354.889
## 3 age==<40yrs 1769 10506.8994 7359.4140 15426.465
## 4 age==>40yrs 111 2183.4543 872.0130 5754.881
## 5 nation==American and Australia 173 708.3688 504.6086 1037.331
## 6 nation==Asia 284 2742.3147 1755.2548 4391.590
## 7 nation==North Africa 1023 3055.2033 2697.4900 3489.333
## 8 nation==Rest of Africa 243 2058.1533 1318.7466 3305.786
## 9 nation==Surinam 64 2386.4544 505.2460 12288.008
## 10 nation==Turkey 93 1739.8592 638.0497 5068.959
popSizestrata_inflated <- stratifyPopsize(modelInflated)
popSizestrata_inflated_report <- popSizestrata_inflated[, cols]
names(popSizestrata_inflated_report) <- cols_custom
popSizestrata_inflated_report## Name Obs Estimated LowerBound UpperBound
## 1 nation==American and Australia 173 516.2432 370.8463 768.4919
## 2 nation==Asia 284 1323.5377 831.1601 2258.9954
## 3 nation==North Africa 1023 2975.8801 2254.7071 4119.3050
## 4 nation==Rest of Africa 243 1033.9753 667.6106 1716.4484
## 5 nation==Surinam 64 354.2236 193.8891 712.4739
## 6 nation==Turkey 93 496.0934 283.1444 947.5309
## 7 gender==female 398 1109.7768 778.7197 1728.7066
## 8 gender==male 1482 5590.1764 3838.4550 8644.0776
## 9 age==<40yrs 1769 6437.8154 4462.3472 9862.2147
## 10 age==>40yrs 111 262.1379 170.9490 492.0347The stratifyPopsize function prepared to handle objects
of the singleRStaticCountData class, accepts three optional
parameters strata, alpha, cov, which are used for
specifying sub-populations, significance levels and the covariance
matrix to be used for computing standard errors. An example of the full
call is presented below.
library(sandwich)
popSizestrataCustom <- stratifyPopsize(
object = basicModel,
strata = ~ gender + age,
alpha = rep(c(0.1, 0.05), each=2),
cov = vcovHC(basicModel, type = "HC4")
)
popSizestrataCustom_report <- popSizestrataCustom[, c(cols, "confLevel")]
names(popSizestrataCustom_report) <- c(cols_custom, "alpha")
popSizestrataCustom_report## Name Obs Estimated LowerBound UpperBound alpha
## 1 gender==female 398 3811.092 2275.6410 6602.168 0.10
## 2 gender==male 1482 8879.261 6261.5109 12930.760 0.10
## 3 age==<40yrs 1769 10506.899 7297.2057 15580.151 0.05
## 4 age==>40yrs 111 2183.454 787.0673 6464.016 0.05We have provided integration with the sandwich (Zeileis, Köll, and Graham
2020) package to correct the variance-covariance matrix in
the
method. In the code we have used the vcovHC method for
singleRStaticCountData class from the
sandwich package, different significance levels for
confidence intervals in each stratum and a formula to specify that we
want estimates for both males and females to be grouped by
nation and age. The strata
parameter can be specified either as:
- a formula with the empty left hand side, as shown in the example
above (e.g.
~ gender * age), - a logical vector with the number of entries equal to the number of
rows in the dataset, in which case only one stratum will be created
(e.g.
netherlandsimmigrant$gender == "male"), - a vector of names of explanatory variables, which will result in
every level of the explanatory variable having its own sub-population
for each variable specified (e.g.
c("gender", "age")), - not supplied at all, in which case strata will correspond to levels of each factor in the data without any interactions (string vectors will be converted to factors for the convenience of the user),
- a (named) list where each element is a logical vector; names of the list will be used to specify variable names in the returned object, for example:
list(
"Stratum 1" = netherlandsimmigrant$gender == "male" &
netherlandsimmigrant$nation == "Suriname",
"Stratum 2" = netherlandsimmigrant$gender == "female" &
netherlandsimmigrant$nation == "North Africa"
)One can also specify plotType = "strata" in the
plot function, which results in a plot with point and CI
estimates of the population size.
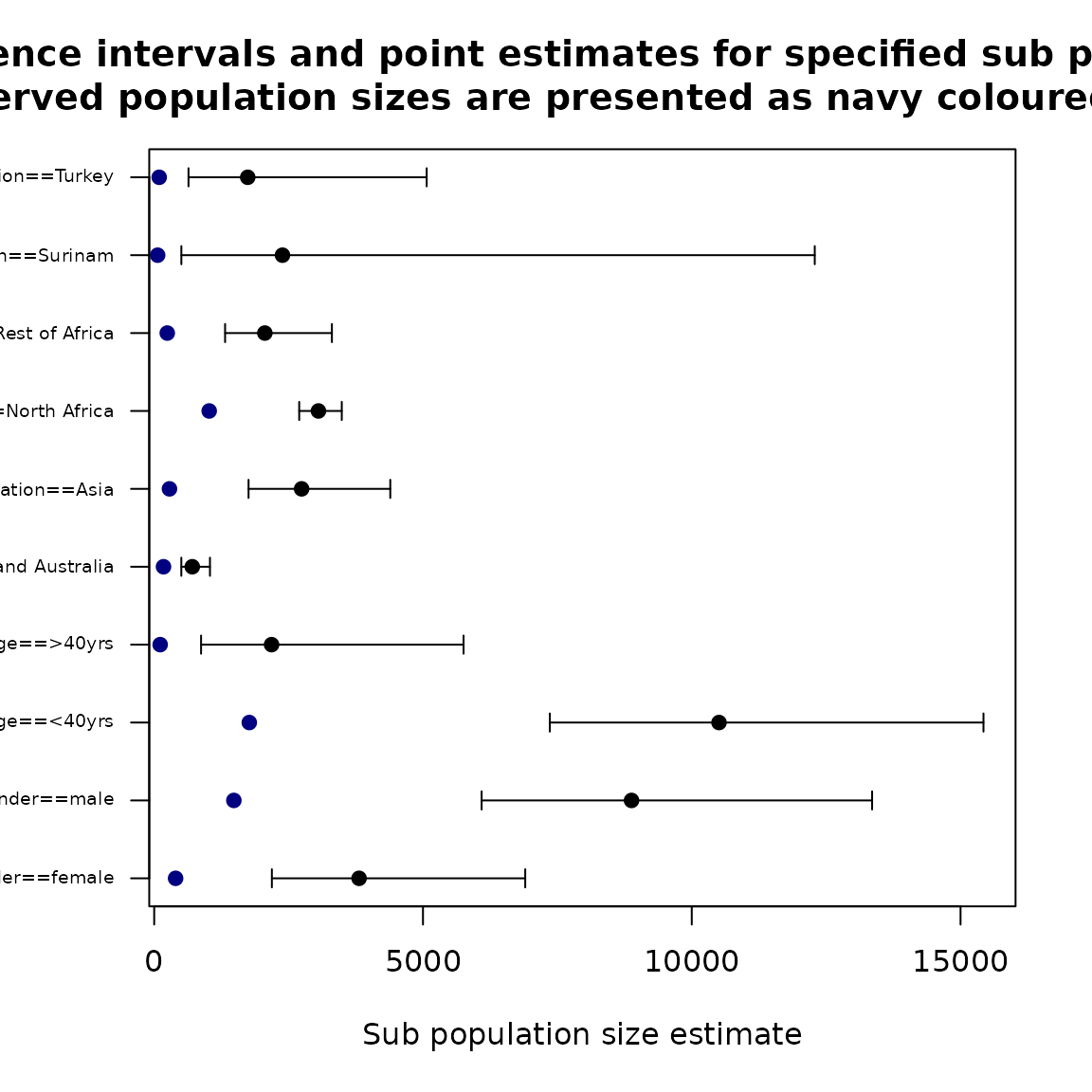
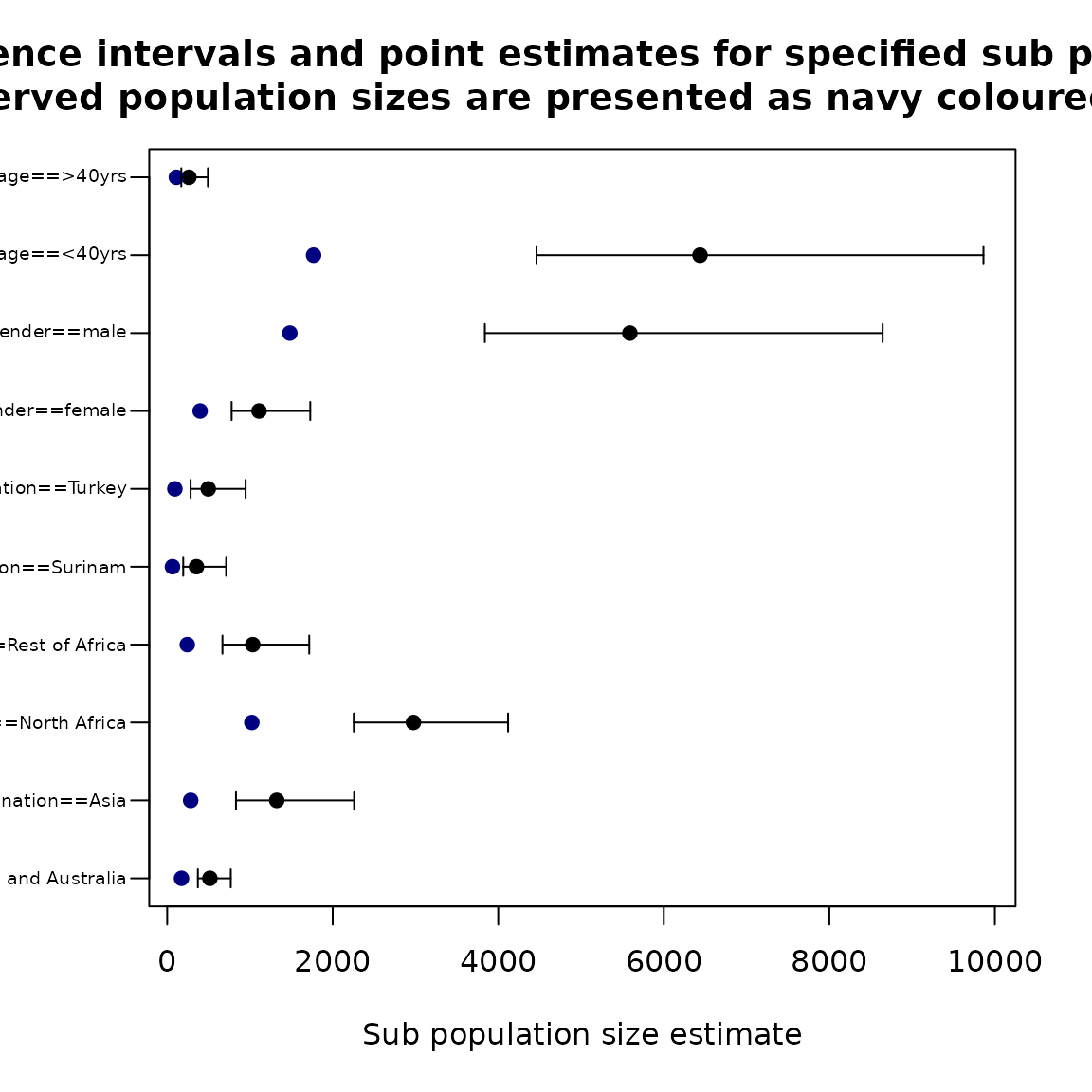
Population size by covariates for ztpoisson (left) and oiztgeom (right) model
Only the logNormal type of confidence interval is used
for plotting since the studentized confidence intervals often result in
negative lower bounds.
Classes and S3Methods
We have created a number of classes. The main ones are:
singleRStaticCountData, singleRfamily, and
supplementary are: popSizeEstResults,
summarysingleRmargin and
summarysingleRStaticCountData, which make it possible to
extract relevant information regarding the population size.
For instance, the popSizeEst function can be used to
extract information about the estimated size of the population as given
below:
(popEst <- popSizeEst(basicModel))## Point estimate: 12690.35
## Variance: 7885812
## 95% confidence intervals:
## lowerBound upperBound
## normal 7186.444 18194.26
## logNormal 8431.275 19718.32and the resulting object popEst of the
popSizeEstResults class contains the following fields:
-
pointEstimate,variance– numerics containing the point estimate and variance of this estimate. -
confidenceInterval– adata.framewith confidence intervals. -
boot– If the bootstrap was performed a numeric vector containing the values from the bootstrap, a character vector with value"No bootstrap performed"otherwise. -
control– acontrolPopVarobject with controls used to obtain the object.
The only explicitly defined method for
popSizeEstResults, summarysingleRmargin and
summarysingleRStaticCountData classes is the
print method, but the former one also accepts
R primitives like coef:
## Estimate Std. Error z value P(>|z|)
## (Intercept) -1.3410661 0.2148870 -6.2407965 4.353484e-10
## gendermale 0.3971793 0.1630155 2.4364504 1.483220e-02
## age>40yrs -0.9746058 0.4082420 -2.3873235 1.697155e-02
## nationAsia -1.0925990 0.3016259 -3.6223642 2.919228e-04
## nationNorth Africa 0.1899980 0.1940007 0.9793677 3.273983e-01
## nationRest of Africa -0.9106361 0.3008092 -3.0272880 2.467587e-03
## nationSurinam -2.3363962 1.0135645 -2.3051282 2.115939e-02
## nationTurkey -1.6753917 0.6027744 -2.7794674 5.444812e-03analogously to glm from stats. The
singleRfamily inherits the family class from
stats and has explicitly defined print and
simulate methods. Example usage is presented below
| Function | Description |
|---|---|
fitted |
It works almost exactly like glm counterparts but
returns more information, namely on fitted values for the truncated and
non-truncated probability distribution; |
logLik |
Compared to glm method, it has the possibility of
returning not just the value of the fitted log-likelihood but also the
entire function (argument type = "function") along with two
first derivatives (argument deriv = 0:2); |
model.matrix |
It has the possibility of returning the matrix defined previously; |
simulate |
It calls the simulate method for the chosen model and
fitted
; |
predict |
It has the possibility of returning either fitted distribution
parameters for each unit (type = "response"), or just
linear predictors (type = "link"), or means of the fitted
distributions of
and
(type = "mean") or the inverse probability weights
(type = "contr"). It is possible to set the
se.fit argument to TRUE in order to obtain
standard errors for each of those by using the
method. Also, it is possible to use a custom covariance matrix for
standard error computation (argument cov); |
redoPopEstimation |
A function that applies all post-hoc procedures that were performed (such as heteroscedastic consistent covariance matrix estimation via countreg) to estimate the population size and standard errors; |
residuals |
Used for obtaining residuals of several types, we refer interested
readers to the manual
?singleRcapture:::residuals.singleRStaticCountData; |
stratifyPopsize, summary |
Compared to the glm class, summary has the possibility
of adding confidence intervals to the coefficient matrix (argument
confint = TRUE) and using a custom covariance matrix
(argument cov = someMatrix); |
plot |
It has been discussed above; |
popSizeEst |
An extractor showcased above; |
cooks.distance |
It works only for single predictor models; |
dfbeta, dfpopsize |
Multi-threading in dfbeta is available and
dfpopsize calls dfbeta if no
dfbeta object was provided in the call; |
bread, estfun, vcovHC |
For (almost) full sandwich compatibility; |
AIC, BIC, extractAIC, family, confint, df.residual, model.frame, hatvalues, nobs, print, sigma, influence, rstudent, rstandard |
These work exactly like glm counterparts. |
set.seed(1234567890)
N <- 10000
gender <- rbinom(N, 1, 0.2)
eta <- -1 + 0.5*gender
counts <- simulate(ztpoisson(), eta = cbind(eta), seed = 1)
summary(data.frame(gender, eta, counts))## gender eta counts
## Min. :0.0000 Min. :-1.0000 Min. :0.0000
## 1st Qu.:0.0000 1st Qu.:-1.0000 1st Qu.:0.0000
## Median :0.0000 Median :-1.0000 Median :0.0000
## Mean :0.2036 Mean :-0.8982 Mean :0.4196
## 3rd Qu.:0.0000 3rd Qu.:-1.0000 3rd Qu.:1.0000
## Max. :1.0000 Max. :-0.5000 Max. :5.0000The full list of explicitly defined methods for
singleRStaticCountData methods is presented in Table
above.
Concluding remarks
In this paper we have introduced the singleRcapture
package for single source capture-recapture models. The package
implement state-of-the-art methods for estimating population size based
on a single data set with multiple counts. The package implements
different methods to account for heterogeneity in capture probabilities,
modelled using covariates, as well as behavioural change, modelled using
one-inflation. We have built the package to facilitate the
implementation of new models using family objects; their
application is exemplified in the Section 7. An example of implementing
a custom family described in Section 8 is presented in replication
materials.
Furthermore, since many R users are familiar with countreg or VGAM packages, we have implemented a lightweight extension called singleRcaptureExtra, available through Github (), which can be used to integrate singleRcapture with these packages.
In future work we plan to implement Bayesian estimation using Stan (e.g. via the brms package; Carpenter et al. (2017), Bürkner (2017)) and for one-inflation models we can use the recent approach proposed by Tuoto, Di Cecco, and Tancredi (2022) and implement our own families using the brms package.
Acknowledgements
The authors’ work has been financed by the National Science Centre in Poland, OPUS 20, grant no. 2020/39/B/HS4/00941.
The authors would like to thank Peter van der Heijden, Maarten Cruyff, Dankmar Böhning, Łukasz Chrostowski and Layna Dennett for useful comments that have helped to improve the functionality of the package. In addition, we would like to thank Marcin Szymkowiak and Tymon Świtalski for their valuable comments that have considerably improved the paper.
Detailed information
The estimatePopsizeFit function
In this section we provide a step-by-step description of how to
prepare data in order to use the estimatePopsizeFit
function, which may be useful to some users, e.g. those wishing to make
modifications to the
estimate or to the bootstrap. In order to show how to apply the function
we will fit a zero truncated geometric model on the data from Böhning et al. (2013) with covariate
dependency:
This would be equivalent to the following esimatePopsize
call:
estimatePopsize(
TOTAL_SUB ~ .,
data = farmsubmission,
model = ztoigeom(),
controlModel(
omegaFormula = ~ 1 + log_size + C_TYPE
)
)- Create a data matrix
- Fill the first
rows with
model.matrixaccording to the specified formula and specify the attributeattr(X, "hwm")that informs the function which elements of the design matrix correspond to which linear predictor (covariates for counts and covariates for one-inflation)
X[1:NROW(farmsubmission), 1:4] <- model.matrix(
~ 1 + log_size + log_distance + C_TYPE,
farmsubmission
)
X[-(1:NROW(farmsubmission)), 5:7] <- model.matrix(
~ 1 + log_distance + C_TYPE,
farmsubmission
)
attr(X, "hwm") <- c(4, 3)- Obtain starting
parameters using the
glm.fitfunction.
start <- glm.fit(
y = farmsubmission$TOTAL_SUB,
x = X[1:NROW(farmsubmission), 1:4],
family = poisson()
)$coefficients
start## [1] -0.82583943 0.33254499 -0.03277732 0.32746933- Use the
estimatePopsizeFitfunction to fit the model assuming a zero-truncated one-inflated geometric distribution as specified in thefamilyargument.
res <- estimatePopsizeFit(
y = farmsubmission$TOTAL_SUB,
X = X,
method = "IRLS",
priorWeights = 1,
family = ztoigeom(),
control = controlMethod(silent = TRUE),
coefStart = c(start, 0, 0, 0),
etaStart = matrix(X %*% c(start, 0, 0, 0), ncol = 2),
offset = cbind(rep(0, NROW(farmsubmission)),
rep(0, NROW(farmsubmission)))
)- Compare our results with those obtained by applying the
stats::optimfunction.
ll <- ztoigeom()$makeMinusLogLike(y = farmsubmission$TOTAL_SUB, X = X)
res2 <- estimatePopsizeFit(
y = farmsubmission$TOTAL_SUB,
X = X,
method = "optim",
priorWeights = 1,
family = ztoigeom(),
coefStart = c(start, 0, 0, 0),
control = controlMethod(silent = TRUE, maxiter = 10000),
offset = cbind(rep(0, NROW(farmsubmission)), rep(0, NROW(farmsubmission)))
)
data.frame(IRLS = round(c(res$beta, -ll(res$beta), res$iter), 4),
optim = round(c(res2$beta, -ll(res2$beta), res2$iter[1]), 4))## IRLS optim
## 1 -2.7845 -2.5971
## 2 0.6170 0.6163
## 3 -0.0646 -0.0825
## 4 0.5346 0.5431
## 5 -3.1745 -0.1504
## 6 0.1281 -0.1586
## 7 -1.0865 -1.0372
## 8 -17278.7613 -17280.1189
## 9 15.0000 1696.0000The default maxiter parameter for "optim"
fitting is
,
but we needed to increase it since the optim does not
converge in
steps and “gets stuck” at a plateau, which results in a lower
log-likelihood value compared to the standard "IRLS".
The above situation is rather typical. While we did not conduct any
formal numerical analyses, it seems that when one attempts to model more
than one parameter of the distribution as covariate dependent
optim algorithms, both "Nelder-Mead" and
"L-BFGS-B" seem to be ill-suited for the task despite being
provided with the analytically computed gradient. This is one of the
reasons why "IRLS" is the default fitting method.
Structure of a family function
In this section we provide details regarding the family
object for the singleRcapture package. This object
contains additional parameters in comparison to the standard
family object from the stats package.
| Function | Description |
|---|---|
makeMinusLogLike |
A factory function for creating the following functions: , , from the vector and the matrix. The deriv argument has possible values in
c(0, 1, 2) that determine which derivative to return; the
default value is 0, which represents the minus
log-likelihood. |
links |
A list with link functions; |
mu.eta, variance |
Functions of linear predictors that return the expected value and
variance. The type argument with 2 possible values
("trunc" and "nontrunc") specifies whether to
return
or
respectively. The deriv argument with values in
c(0, 1, 2) is used for indicating the derivative with
respect to the linear predictors, which is used for providing standard
errors in the predict method. |
family |
A string that specifies the model name; |
valideta, validmu |
For now, it only returns TRUE. In the near future, it
will be used to check whether applied linear predictors are valid (i.e.,
are transformed into some elements of the parameter space subjected to
the inverse link function). |
funcZ, Wfun |
Functions that create pseudo residuals and working weights used in the IRLS algorithm; |
devResids |
A function that returns deviance residuals given a vector of prior weights of linear predictors and the response vector; |
pointEst, popVar |
Functions that return the point estimate for the population size and
analytic estimation of its variance given prior weights of linear
predictors and, in the latter case, also estimates of
and
matrix. There is an additional boolean parameter contr in
the former function, which, if set to TRUE, returns the
contribution of each unit. |
etaNames |
Names of linear predictors; |
densityFunction |
A function that returns the value of PMF at values of x
given linear predictors. The type argument specifies
whether to return
or
; |
simulate |
A function that generates values of a dependent vector given linear predictors; |
getStart |
An expression for generating starting points; |
Implementing a custom singleRcapture family function
Suppose we want to implement a very specific zero truncated family function in the singleRcapture, which corresponds to the following “untruncated” distribution:
with being dependent on covariates.
Below we provide a possible way of implementing the above model, with
lambda, pi meaning
.
We provide a simple example that shows that the proposed approach works
as expected.
myFamilyFunction <- function(lambdaLink = c("logit", "cloglog", "probit"),
piLink = c("logit", "cloglog", "probit"),
...) {
if (missing(lambdaLink)) lambdaLink <- "logit"
if (missing(piLink)) piLink <- "logit"
links <- list()
attr(links, "linkNames") <- c(lambdaLink, piLink)
lambdaLink <- switch(lambdaLink,
"logit" = singleRcapture:::singleRinternallogitLink,
"cloglog" = singleRcapture:::singleRinternalcloglogLink,
"probit" = singleRcapture:::singleRinternalprobitLink
)
piLink <- switch(piLink,
"logit" = singleRcapture:::singleRinternallogitLink,
"cloglog" = singleRcapture:::singleRinternalcloglogLink,
"probit" = singleRcapture:::singleRinternalprobitLink
)
links[1:2] <- c(lambdaLink, piLink)
mu.eta <- function(eta, type = "trunc", deriv = FALSE, ...) {
pi <- piLink(eta[, 2], inverse = TRUE) / 2
lambda <- lambdaLink(eta[, 1], inverse = TRUE) / 2
if (!deriv) {
switch (type,
"nontrunc" = pi + 2 * lambda,
"trunc" = 1 + lambda / (pi + lambda)
)
} else {
# Only necessary if one wishes to use standard errors in predict method
switch (type,
"nontrunc" = {
matrix(c(2, 1) * c(
lambdaLink(eta[, 1], inverse = TRUE, deriv = 1) / 2,
piLink(eta[, 2], inverse = TRUE, deriv = 1) / 2
), ncol = 2)
},
"trunc" = {
matrix(c(
pi / (pi + lambda) ^ 2,
-lambda / (pi + lambda) ^ 2
) * c(
lambdaLink(eta[, 1], inverse = TRUE, deriv = 1) / 2,
piLink(eta[, 2], inverse = TRUE, deriv = 1) / 2
), ncol = 2)
}
)
}
}
variance <- function(eta, type = "nontrunc", ...) {
pi <- piLink(eta[, 2], inverse = TRUE) / 2
lambda <- lambdaLink(eta[, 1], inverse = TRUE) / 2
switch (type,
"nontrunc" = pi * (1 - pi) + 4 * lambda * (1 - lambda - pi),
"trunc" = lambda * (1 - lambda) / (pi + lambda)
)
}
Wfun <- function(prior, y, eta, ...) {
pi <- piLink(eta[, 2], inverse = TRUE) / 2
lambda <- lambdaLink(eta[, 1], inverse = TRUE) / 2
G01 <- ((lambda + pi) ^ (-2)) * piLink(eta[, 2], inverse = TRUE, deriv = 1) *
lambdaLink(eta[, 1], inverse = TRUE, deriv = 1) * prior / 4
G00 <- ((lambda + pi) ^ (-2)) - (pi ^ (-2)) - lambda / ((lambda + pi) * (pi ^ 2))
G00 <- G00 * prior * (piLink(eta[, 2], inverse = TRUE, deriv = 1) ^ 2) / 4
G11 <- ((lambda + pi) ^ (-2)) - (((lambda + pi) * lambda) ^ -1)
G11 <- G11 * prior * (lambdaLink(eta[, 1], inverse = TRUE, deriv = 1) ^ 2) / 4
matrix(
-c(G11, # lambda
G01, # mixed
G01, # mixed
G00 # pi
),
dimnames = list(rownames(eta), c("lambda", "mixed", "mixed", "pi")),
ncol = 4
)
}
funcZ <- function(eta, weight, y, prior, ...) {
pi <- piLink(eta[, 2], inverse = TRUE) / 2
lambda <- lambdaLink(eta[, 1], inverse = TRUE) / 2
weight <- weight / prior
G0 <- (2 - y) / pi - ((lambda + pi) ^ -1)
G1 <- (y - 1) / lambda - ((lambda + pi) ^ -1)
G1 <- G1 * lambdaLink(eta[, 1], inverse = TRUE, deriv = 1) / 2
G0 <- G0 * piLink(eta[, 2], inverse = TRUE, deriv = 1) / 2
uMatrix <- matrix(c(G1, G0), ncol = 2)
weight <- lapply(X = 1:nrow(weight), FUN = function (x) {
matrix(as.numeric(weight[x, ]), ncol = 2)
})
pseudoResid <- sapply(X = 1:length(weight), FUN = function (x) {
#xx <- chol2inv(chol(weight[[x]])) # less computationally demanding
xx <- solve(weight[[x]]) # more stable
xx %*% uMatrix[x, ]
})
pseudoResid <- t(pseudoResid)
dimnames(pseudoResid) <- dimnames(eta)
pseudoResid
}
minusLogLike <- function(y, X, offset,
weight = 1,
NbyK = FALSE,
vectorDer = FALSE,
deriv = 0,
...) {
y <- as.numeric(y)
if (is.null(weight)) {
weight <- 1
}
if (missing(offset)) {
offset <- cbind(rep(0, NROW(X) / 2), rep(0, NROW(X) / 2))
}
if (!(deriv %in% c(0, 1, 2)))
stop("Only score function and derivatives up to 2 are supported.")
deriv <- deriv + 1
switch (deriv,
function(beta) {
eta <- matrix(as.matrix(X) %*% beta, ncol = 2) + offset
pi <- piLink(eta[, 2], inverse = TRUE) / 2
lambda <- lambdaLink(eta[, 1], inverse = TRUE) / 2
-sum(weight * ((2 - y) * log(pi) + (y - 1) * log(lambda) - log(pi + lambda)))
},
function(beta) {
eta <- matrix(as.matrix(X) %*% beta, ncol = 2) + offset
pi <- piLink(eta[, 2], inverse = TRUE) / 2
lambda <- lambdaLink(eta[, 1], inverse = TRUE) / 2
G0 <- (2 - y) / pi - ((lambda + pi) ^ -1)
G1 <- (y - 1) / lambda - ((lambda + pi) ^ -1)
G1 <- G1 * weight * lambdaLink(eta[, 1], inverse = TRUE, deriv = 1) / 2
G0 <- G0 * weight * piLink(eta[, 2], inverse = TRUE, deriv = 1) / 2
if (NbyK) {
XX <- 1:(attr(X, "hwm")[1])
return(cbind(as.data.frame(X[1:nrow(eta), XX]) * G1,
as.data.frame(X[-(1:nrow(eta)), -XX]) * G0))
}
if (vectorDer) {
return(cbind(G1, G0))
}
as.numeric(c(G1, G0) %*% X)
},
function (beta) {
lambdaPredNumber <- attr(X, "hwm")[1]
eta <- matrix(as.matrix(X) %*% beta, ncol = 2) + offset
pi <- piLink(eta[, 2], inverse = TRUE) / 2
lambda <- lambdaLink(eta[, 1], inverse = TRUE) / 2
res <- matrix(nrow = length(beta), ncol = length(beta),
dimnames = list(names(beta), names(beta)))
# pi^2 derivative
dpi <- (2 - y) / pi - (lambda + pi) ^ -1
G00 <- ((lambda + pi) ^ (-2)) - (2 - y) / (pi ^ 2)
G00 <- t(as.data.frame(X[-(1:(nrow(X) / 2)), -(1:lambdaPredNumber)] *
(G00 * ((piLink(eta[, 2], inverse = TRUE, deriv = 1) / 2) ^ 2) +
dpi * piLink(eta[, 2], inverse = TRUE, deriv = 2) / 2) * weight)) %*%
as.matrix(X[-(1:(nrow(X) / 2)), -(1:lambdaPredNumber)])
# mixed derivative
G01 <- (lambda + pi) ^ (-2)
G01 <- t(as.data.frame(X[1:(nrow(X) / 2), 1:lambdaPredNumber]) *
G01 * (lambdaLink(eta[, 1], inverse = TRUE, deriv = 1) / 2) *
(piLink(eta[, 2], inverse = TRUE, deriv = 1) / 2) * weight) %*%
as.matrix(X[-(1:(nrow(X) / 2)), -(1:lambdaPredNumber)])
# lambda^2 derivative
G11 <- ((lambda + pi) ^ (-2)) - (y - 1) / (lambda ^ 2)
dlambda <- (y - 1) / lambda - ((lambda + pi) ^ -1)
G11 <- t(as.data.frame(X[1:(nrow(X) / 2), 1:lambdaPredNumber] *
(G11 * ((lambdaLink(eta[, 1], inverse = TRUE, deriv = 1) / 2) ^ 2) +
dlambda * lambdaLink(eta[, 1], inverse = TRUE, deriv = 2) / 2) * weight)) %*%
X[1:(nrow(X) / 2), 1:lambdaPredNumber]
res[-(1:lambdaPredNumber), -(1:lambdaPredNumber)] <- G00
res[1:lambdaPredNumber, 1:lambdaPredNumber] <- G11
res[1:lambdaPredNumber, -(1:lambdaPredNumber)] <- t(G01)
res[-(1:lambdaPredNumber), 1:lambdaPredNumber] <- G01
res
}
)
}
validmu <- function(mu) {
(sum(!is.finite(mu)) == 0) && all(0 < mu) && all(2 > mu)
}
# this is optional
devResids <- function(y, eta, wt, ...) {
0
}
pointEst <- function (pw, eta, contr = FALSE, ...) {
pi <- piLink(eta[, 2], inverse = TRUE) / 2
lambda <- lambdaLink(eta[, 1], inverse = TRUE) / 2
N <- pw / (lambda + pi)
if(!contr) {
N <- sum(N)
}
N
}
popVar <- function (pw, eta, cov, Xvlm, ...) {
pi <- piLink(eta[, 2], inverse = TRUE) / 2
lambda <- lambdaLink(eta[, 1], inverse = TRUE) / 2
bigTheta1 <- -pw / (pi + lambda) ^ 2 # w.r to pi
bigTheta1 <- bigTheta1 * piLink(eta[, 2], inverse = TRUE, deriv = 1) / 2
bigTheta2 <- -pw / (pi + lambda) ^ 2 # w.r to lambda
bigTheta2 <- bigTheta2 * lambdaLink(eta[, 1], inverse = TRUE, deriv = 1) / 2 # w.r to lambda
bigTheta <- t(c(bigTheta2, bigTheta1) %*% Xvlm)
f1 <- t(bigTheta) %*% as.matrix(cov) %*% bigTheta
f2 <- sum(pw * (1 - pi - lambda) / ((pi + lambda) ^ 2))
f1 + f2
}
dFun <- function (x, eta, type = c("trunc", "nontrunc")) {
if (missing(type)) type <- "trunc"
pi <- piLink(eta[, 2], inverse = TRUE) / 2
lambda <- lambdaLink(eta[, 1], inverse = TRUE) / 2
switch (type,
"trunc" = {
(pi * as.numeric(x == 1) + lambda * as.numeric(x == 2)) / (pi + lambda)
},
"nontrunc" = {
(1 - pi - lambda) * as.numeric(x == 0) +
pi * as.numeric(x == 1) + lambda * as.numeric(x == 2)
}
)
}
simulate <- function(n, eta, lower = 0, upper = Inf) {
pi <- piLink(eta[, 2], inverse = TRUE) / 2
lambda <- lambdaLink(eta[, 1], inverse = TRUE) / 2
CDF <- function(x) {
ifelse(x == Inf, 1,
ifelse(x < 0, 0,
ifelse(x < 1, 1 - pi - lambda,
ifelse(x < 2, 1 - lambda, 1))))
}
lb <- CDF(lower)
ub <- CDF(upper)
p_u <- stats::runif(n, lb, ub)
sims <- rep(0, n)
cond <- CDF(sims) <= p_u
while (any(cond)) {
sims[cond] <- sims[cond] + 1
cond <- CDF(sims) <= p_u
}
sims
}
getStart <- expression(
if (method == "IRLS") {
etaStart <- cbind(
family$links[[1]](mean(observed == 2) * (1 + 0 * (observed == 2))), # lambda
family$links[[2]](mean(observed == 1) * (1 + 0 * (observed == 1))) # pi
) + offset
} else if (method == "optim") {
init <- c(
family$links[[1]](weighted.mean(observed == 2, priorWeights) * 1 + .0001),
family$links[[2]](weighted.mean(observed == 1, priorWeights) * 1 + .0001)
)
if (attr(terms, "intercept")) {
coefStart <- c(init[1], rep(0, attr(Xvlm, "hwm")[1] - 1))
} else {
coefStart <- rep(init[1] / attr(Xvlm, "hwm")[1], attr(Xvlm, "hwm")[1])
}
if ("(Intercept):pi" %in% colnames(Xvlm)) {
coefStart <- c(coefStart, init[2], rep(0, attr(Xvlm, "hwm")[2] - 1))
} else {
coefStart <- c(coefStart, rep(init[2] / attr(Xvlm, "hwm")[2], attr(Xvlm, "hwm")[2]))
}
}
)
structure(
list(
makeMinusLogLike = minusLogLike,
densityFunction = dFun,
links = links,
mu.eta = mu.eta,
valideta = function (eta) {TRUE},
variance = variance,
Wfun = Wfun,
funcZ = funcZ,
devResids = devResids,
validmu = validmu,
pointEst = pointEst,
popVar = popVar,
family = "myFamilyFunction",
etaNames = c("lambda", "pi"),
simulate = simulate,
getStart = getStart,
extraInfo = c(
mean = "pi / 2 + lambda",
variance = paste0("(pi / 2) * (1 - pi / 2) + 2 * lambda * (1 - lambda / 2 - pi / 2)"),
popSizeEst = "(1 - (pi + lambda) / 2) ^ -1",
meanTr = "1 + lambda / (pi + lambda)",
varianceTr = paste0("lambda * (1 - lambda / 2) / (pi + lambda)")
)
),
class = c("singleRfamily", "family")
)
}A quick tests shows us that this implementation in fact works:
set.seed(123)
Y <- simulate(
myFamilyFunction(lambdaLink = "logit", piLink = "logit"),
nsim = 1000, eta = matrix(0, nrow = 1000, ncol = 2),
truncated = FALSE
)
mm <- estimatePopsize(
formula = Y ~ 1,
data = data.frame(Y = Y[Y > 0]),
model = myFamilyFunction(lambdaLink = "logit",
piLink = "logit"),
# the usual observed information matrix
# is ill-suited for this distribution
controlPopVar = controlPopVar(covType = "Fisher")
)
summary(mm)##
## Call:
## estimatePopsize.default(formula = Y ~ 1, data = data.frame(Y = Y[Y >
## 0]), model = myFamilyFunction(lambdaLink = "logit", piLink = "logit"),
## controlPopVar = controlPopVar(covType = "Fisher"))
##
## Pearson Residuals:
## Min. 1st Qu. Median Mean 3rd Qu. Max.
## -0.8198 -0.8198 0.8099 0.0000 0.8099 0.8099
##
## Coefficients:
## -----------------------
## For linear predictors associated with: lambda
## Estimate Std. Error z value P(>|z|)
## (Intercept) 0.01217 0.20253 0.06 0.952
## -----------------------
## For linear predictors associated with: pi
## Estimate Std. Error z value P(>|z|)
## (Intercept) -0.01217 0.08926 -0.136 0.892
##
## AIC: 687.4249
## BIC: 695.8259
## Residual deviance: 0
##
## Log-likelihood: -341.7124 on 984 Degrees of freedom
## Number of iterations: 2
## -----------------------
## Population size estimation results:
## Point estimate 986
## Observed proportion: 50% (N obs = 493)
## Std. Error 70.30092
## 95% CI for the population size:
## lowerBound upperBound
## normal 848.2127 1123.787
## logNormal 866.3167 1144.053
## 95% CI for the share of observed population:
## lowerBound upperBound
## normal 43.86951 58.12221
## logNormal 43.09241 56.90759where the link functions, such as
singleRcapture:::singleRinternalcloglogLink, are just
internal functions in singleRcapture that compute link
functions, their inverses and derivatives of both links and inverse
links up to the third order:
singleRcapture:::singleRinternalcloglogLink## function (x, inverse = FALSE, deriv = 0)
## {
## deriv <- deriv + 1
## if (isFALSE(inverse)) {
## res <- switch(deriv, log(-log(1 - x)), -1/((1 - x) *
## log(1 - x)), -(1 + log(1 - x))/((x - 1)^2 * log(1 -
## x)^2), (2 * log(1 - x)^2 + 3 * log(1 - x) + 2)/(log(1 -
## x)^3 * (x - 1)^3))
## }
## else {
## res <- switch(deriv, 1 - exp(-exp(x)), exp(x - exp(x)),
## (1 - exp(x)) * exp(x - exp(x)), (exp(2 * x) - 3 *
## exp(x) + 1) * exp(x - exp(x)))
## }
## res
## }
## <bytecode: 0x55f812f82f50>
## <environment: namespace:singleRcapture>One could, of course, include the code for computing them manually.